Revenue forecasting is an essential component of financial planning for state and local governments. Forecasts directly influence budgeting, policy decisions, and overall economic stability. Traditionally, debates around the “best” forecasting techniques have revolved around comparing classical statistical methods with modern machine learning techniques. However, new insights from Sarah E. Larson and Michael Overton’s (2024) research challenge this narrative, emphasizing a less-discussed yet critical aspect: data preprocessing.
Shifting the Focus: Why Preprocessing Matters More
The study explored various revenue forecasting methods for sales tax revenues, comparing traditional techniques like ARIMA and exponential smoothing with advanced machine learning approaches such as k-nearest neighbors (KKNN) and extreme gradient boosting (XGBOOST). Surprisingly, the research found that the choice of forecasting model—while important—had less impact on accuracy compared to how data was prepared beforehand.
Preprocessing refers to cleaning data prior to analysis and includes steps like detrending data, seasonally adjusting values, or applying transformations like logarithms or the inverse hyperbolic sine (IHS). These steps address inherent challenges in time-series data, such as seasonality and long-term trends that if left unaddressed will bias forecasts.
Key Findings: Consistency Across Time Intervals
Larson and Overton analyzed over 16 years of monthly, quarterly, and annual sales tax data from Texas cities. The findings underscored that:
Preprocessing Steps Drive Accuracy: Transformations like IHS and logarithms consistently improved accuracy across all forecasting intervals. These methods normalized the data and reduced the influence of outliers.
Time-Series Characteristics Matter: Adjustments for seasonality and trends significantly enhanced model performance, particularly for monthly and quarterly forecasts.
Model Selection Is Secondary: While machine learning models like KKNN and XGBOOST excelled in certain contexts, benchmark methods, such as the Drift and Seasonal Naïve models, often performed comparably after robust preprocessing.
Machine Learning vs. Traditional Methods
The promise of machine learning often lies in its ability to uncover complex patterns in large datasets. Yet, Larson and Overton’s findings reveal that machine learning models are not a magic solution. Their accuracy can be hindered if the input data lacks proper preprocessing, leading to overfitting or errors from unaddressed trends and seasonality.
Interestingly, when applied to well-preprocessed data, traditional models matched or exceeded the performance of advanced algorithms. This result calls into question the rush to adopt sophisticated tools without first mastering fundamental data preparation practices.
Practical Implications for Forecasters
For practitioners and policymakers, the message is clear: the focus should shift from “which model to use” to “how to preprocess the data effectively.” Key recommendations include:
Prioritize Normalization: Methods like IHS and logarithmic transformations should be routine, especially for data with high variability.
Account for Trends and Seasonality: Detrending and seasonal adjustments should be standard practices for time-series analysis.
Match Techniques to Time Intervals: Different preprocessing steps and models work better for monthly, quarterly, or yearly forecasts. Tailoring the approach to the data interval ensures better results.
Be Cautious with Inflation Adjustments: While important, inflation adjustments do not always enhance accuracy and should be applied judiciously.
The Road Ahead: Rethinking Forecasting Research
Larson and Overton’s study challenges entrenched assumptions in the field. It suggests that future research should explore the interplay between preprocessing and forecasting methods more deeply rather than fixating solely on model innovation. Additionally, replicating this study in other states or revenue contexts could further validate its findings and refine best practices.
References
Larson, S., & Overton, M. (2024). Modeling Approach Matters, But Not as Much as Preprocessing: Comparison of Machine Learning and Traditional Revenue Forecasting Techniques. Public Finance Journal, 1(1), 29–48. https://doi.org/10.59469/pfj.2024.8
In the digital age, predicting the outcome of U.S. elections has become both an art and a science, captivating millions of Americans every election cycle. Although political scientists have a long history of attempting to predict and explain elections, Nate Silver’s FiveThirtyEight blog brought these models to many more Americans in 2008. That year, Silver predicted an Obama victory in the popular vote by 6.1 percentage points and an end electoral college vote of around 350, which ended up just below Obama’s actual margin of 7.3 percentage points and 365 electoral college votes. Silver’s website also garnered over 5 million visits on election day.[1]
By our count, there are at least seven of these forecasting models including models developed by The Economist and ABC News. Each of these models combines a wide array of data to generate a specific quantitative prediction for their respective forecasts (note that the way these predictions are displayed varies). In this blog we outline how these different forecast models work, the type of information they use, and the assumptions they make (implicitly or explicitly).
It is worth noting what forecast models are not. Forecast models do not include polling aggregators like The New York Times and Real Clear Politics’s aggregators. Unlike forecast models, polling aggregators are designed to provide a snapshot of the election as it stands. They do not explicitly project forward who is likely to win. Forecast models are also different than the expert intuition models such as Keys to the White House, Sabato’s Crystal Ball, and Cook Political Report. The output of these expert models varies significantly, with some focusing on state-level predictions and some overall election-level predictions. The inputs to these models also vary significantly and are often not explicitly stated. Instead, an expert uses different cues to identify who they think will win based on past experience. To clarify the landscape of election forecasting, it’s essential to distinguish between the different types of models available. Table 1 outlines the primary categories, detailing their methodologies and providing examples of each.
Table 1: Types of Election Models
TYPE
DESCRIPTIONS
EXAMPLES
Forecasting
Utilizes a combination of polling data and other quantitative metrics to produce specific predictions on election outcomes. These models often incorporate statistical techniques to adjust for biases and trends.
The Economist, 538, Decision Desk HQ
Polling Aggregation
Aggregates multiple polls to provide a current snapshot of the race without projecting future outcomes. These aggregators typically average poll results to show the present state of voter preferences.
New York Times, Real Clear Polling
Expert Intuition
Relies on the qualitative and quantitative insights of political analysts and experts. These models depend on the experts’ experience and judgment to assess various indicators and predict likely winners.
Keys to the White House, Sabato’s Crystal Ball, Cook Political Report
What is in a forecasting model?
Polling
To better understand election forecasts we collected information from a variety of prominent forecasts; the full list of models examined is in Table 2. All these models incorporate polling data, but a critical initial decision is determining which polls to include or exclude. Surprisingly, most election models lack explicit criteria for poll inclusion. FiveThirtyEight provides the most comprehensive rules, requiring methodological transparency and that pollsters meet ethical standards as outlined by the American Association for Public Opinion Research (AAPOR). Silver Bulletin uses similar rules (although has a slightly different list of “banned” pollsters). In addition, several (JHK and possibly The Economist) rely on FiveThirtyEight’s database of polls. In contrast to this, Race to the White House and Decision Desk both maintain their own polling database with very little transparency about how polls are added to it. Overall though it appears that in many cases the polling data used is the same across the different forecasts.
Although the forecasts start with much of the same polling data they immediately begin to diverge significantly in how they deal with the variation within pollsters. Polls can vary in many ways including the size of the sample, their modality (phones with a live caller, phones with automated messages, online, etc.), how they invite individuals to participate (phone, text, online panel), decisions made on how to weight the sample, and the funding of the poll. Most forecasts attempt to correct polls for these different effects by estimating models of the polls to calculate the bias induced by different polling decisions. After corrections are made the polls are averaged with several forecasts (FiveThirtyEight, Silver Bulletin, and JHK) weighting each poll as a function of the sample size, recency, and FiveThirtyEight pollster rating. One interesting exception is DecisionDesk, which simply fits a cubic spline to the polls.
Fundamentals
Nearly every forecasting model incorporates non-polling data, referred to as ‘fundamentals,’ which aim to capture the underlying factors influencing the election. This can include a wide array of data such as economic indicators, historical partisan lean, and consumer outlook. There is a significant variation in both how transparent models are with their fundamentals and which fundamentals they include. For example, FiveThirtyEight includes 11 different economic variables and a handful of political variables at the state or national level. Race to the White House in contrast includes 10 economic variables, that often overlap with FiveThirtyEight’s. For example, both include the University of Michigan’s consumer sentiment index and inflation measured by the CPI. They also have small differences such as the Race To the White House including the NASDAQ Composite Index while FiveThirtyEight includes the S&P 500 and FiveThirtyEight including slightly different measures of income. There are bigger differences for political variables. FiveThirtyEight mainly focuses on historic partisan lean while also accounting for specifics of the candidate (incumbency and home state advantages). Race to the White House though also pulls fundraising data and information from special elections.
Many of the other remaining models are not as transparent in the data that is included. The Economist provides important details in the process used to select the variables at the national level[2] which they say identifies a set of variables that are similar to Alan Abramowitz’s Time for Change Model (which includes presidential approval, change in real GNP, and an indicator for incumbency). Other models often only identify that they have created an “economic index” though they often go into deep detail on assumptions over incumbency. It isn’t clear the degree that different measures of economic indicators matter, although The Economist’s approach does demonstrate that there are principled ways to build these models of fundamentals.
State Correlation
Another important component for forecasts are the state-level estimates. As should be clear, the US presidential election is not decided by popular vote but is instead decided by electors tied to specific states. This means that the national level popular vote is only somewhat informative over who will win (see Hilary Clinton in 2016 and Al Gore in 2000). State-level estimates are then necessary, but state-level polling is significantly less common than national-level polls. In response to this most forecasts attempt to connect polling from one state to polling from other states (as well as to national polls). They do this by incorporating state-level correlations either explicitly in their model of polls or in their simulations. Most forecasts use ancillary data to estimate similarities across states. For example, Decision Desk HQ uses a “comprehensive set of demographic, geographic, and political variables” to identify 10 regions. In their end simulations, each region has its own error which moves state-level polls in the same direction for states in those regions. Others, like The Economist, directly incorporate state-level relationships into their model. Similar to Decision Desk HQ though The Economist also relies on a set of demographic and political factors to measure how similar two states are.
Table 2: Forecast Models
Publisher
Poll Sources
Economic Fundamentals
State Correlation
24cast
Relies on 538
Yes
No
Cnalysis
Relies on JHK average
No
?
Decision Desk HQ
No explicit guidelines
No
Yes
FiveThirtyEight
Specific requirements
Yes
Yes
JHK Forecast
Relies on 538
Yes
Yes
Princeton Election Consortium
Relies on 538
No
No
Race to the WH
No explicit guidelines
Yes
No
Silver Bulletin
Specific requirements
?
Yes
The Economist
No explicit guidelines, possibly relies on 538
Yes
Yes
Conclusion
Election forecasting models have grown both in complexity and profitability over the past two decades. This growth presents a paradox: as models become more sophisticated, the need for transparency increases to understand the underlying assumptions and methodologies. Currently, FiveThirtyEight is the only model among those discussed that openly shares a significant portion of its data, model structure, and outputs. The reason is likely a function of the second trend. Other models like Silver Bulletin, Decision Desk HQ, and The Economist operate behind paywalls, which, while understandable for funding purposes, hinder comparative analysis and public understanding. Given that it is unlikely for these paywalls to fall it would be a positive step for the people behind these models to commit to making their work public after the election. This is a big ask. It is likely that some of these models will be proven wrong, but we can better learn from them only if their details are made public.
Join Kevin Reuning, Center for Analytics and Data Science Faculty Fellow and assistant professor in Political Science, as he leads an activity-driven exploration of the (sometimes hidden) connections that link us as a society.
Over three consecutive Wednesday evenings in McVey 168, Reuning will help the audience take their knowledge of R and data analysis as it pertains to more traditional data sets and apply it to the interconnected web that is the foundation of a social networking modelling.
March 6th: Introduction to network terminology and data
March 13th: Visualizing networks
March 20th: Calculating basic network statistics
This entry in the CADS Faculty Fellow Boot Camp Series presumes at least some level of R proficiency and working knowledge of basic data analysis principles. Due to the interactive nature of the exploration, please bring your laptop.
The Center for Analytics and Data Science is proud to be able to bring unique views into the arena of data science through its Faculty Fellow program. Thanks to the wide variety of talent offered by these gifted academics, CADS is able to provide examples of data science principles as they apply to the research of an array of disciplines. We thank all of our Faculty Fellows for their hard work and willingness to share.
If you have a topic that you would like to see covered as part of the Faculty Fellows Bootcamp Series, or any other question please contact the Center for Analytics and Data Science at [email protected]
This blog post has been written by Dr. Jing Zhang, adopted from research she presented with Dr. Thomas Fisher and Qi He, Miami University masters student.
Linear
regression is likely to be the first statistical modeling tool that many of you
learn in your coursework, no matter you are a data science and statistics
major, math and stat major, analytics co-major or data analytics major. It is
still a popular modeling tool because it is easy to implement
mathematically/computationally, and the model findings are intuitive to
interpret. However, it also suffers from the lack of flexibility due to all the
important model assumptions that are possibly problematic in real analysis:
normality of responses, independence among observations, linear relation
between the response and group of predictors. When outliers exists in the data,
or the predictors are highly correlated with each other, the model findings
will be distorted and therefore misleading in decision-making. What’s more, it
suffers more in the “big-data” era.
A
real “BIG” data set would be too big to hold in a single computer’s memory,
while a “big” data simply mean that the data set is so big that the traditional
statistical analysis tools, such as the linear regression model, would be too
time consuming to implement, and suffers more from the rounding errors in
computation.
Big data sets often have many variables, i.e. a lot of predictors in the linear regression model (i.e., large “p”), in addition to the large number of data observations (i.e., large “n”). Therefore, we need to select the “true” signals among lots “noises” when fitting any statistical models to such a data set, in another word, we often need to conduct “variable selection.” We wish to speed up the model fitting, variable selection and prediction in analysis of big data sets, yet with relatively simple modeling tools, such as linear regression.
Popular
choices involves subsampling the big data and focusing on the analysis of the
subset of information. For
example, bags of little bootstraps (Kleiner et al. 2014) suggested selecting a
random sample of the data and then using bootstrap on the selected sample to
recover the original size of the data in the following analysis, which would
effectively reduce the memory required in the data storage and analysis, and
help in the “BIG” data case.
In addition to random selection,
researchers also suggested using sampling weights based on the features of data
observation, such as the leverage values (Ma and Sun, 2014) to retain the
feature of the data set as much as we can when the subsampling has to be done.
Alternatively, the “divide and
conquer” idea has also been popular: big
data are split into multiple blocks of smaller sample size without overlap and
the analysis results of each block are then aggregated to obtain the final
estimated model and predictions (Chen and Xie, 2014). This idea utilizes all the information in the data sets.
In a CADS funded project, we explore the revamping of linear regression in big
data predictive modeling using a similar “divide and resample” idea, via a
two-stage algorithm. Firstly, we combine the least absolute shrinkage and
selection operator (LASSO) (Tibshirani, 1996), which helps select the relevant
features; and the divide and conquer approach, which helps deal with the large
sample size, in the variable selection stage, with products being the selected
relevant features in the big data with high dimension. Secondly, in the
prediction stage, with the selected features, when the data are of extremely
high sample size, we subsample the data multiple times, refit the model with
selected features to each subsample, and then aggregate the predictions based
on the analysis of each subsample. When the data are sizable but the chosen
model can still be fitted with reasonable computing cost, predictions are obtained
directly by refitting the model with selected features to the complete data. Here
is the detailed description of the algorithm:
Step 1. Partition a large data set
of sample size into blocks, each with sample size . The samples are randomly assigned to
these blocks.
Step 2. Conduct variable selection
with LASSO in each block individually.
Step 3. Use “majority voting” to
select the variables, i.e, if we have a “majority” of blocks end up selecting a
predictor variable in the analysis, this predictor is retained.
Step 4. When “” is large but still can
be handled as one piece,
refit the model with selected variables to the
original data and predict based on the
parameter estimates. When “” is too large to be
handled as one piece, randomly select multiple subsample from the original data
and refit the model with selected variables on the subsamples, then aggregate
the predictions based on the models fitted to different subsamples (e.g. mean
or median of the predicted values).
Simulation
studies are often used to evaluate the performance of statistical models,
methods and algorithms. In a simulation study, we are able to simulate
“plausible” data from a known probabilistic framework with pre-picked values of
model parameters. Then the proposed statistical analysis method would be
implemented to fit the simulated data, and the analysis findings, such as the
estimated parameters, predicted responses of a holdout set, would be compared
with the true values we know in the simulation. When many such plausible data
sets are simulated and analyzed, we are able to empirically evaluate the
performance of the proposed methods through the discrepancy between the model
findings and true values. In this project, we conducted a simulation study to
help make important decisions on key components of the proposed algorithm,
including the number of blocks we divide the complete data into, and how to
decide “majority” in the variable selection step. This simulation study was
also designed such that we are able to evaluate the impact of multicolinearity
(highly correlated predictors) and effect size (strength of the linear relation
between response and predictors) on the performance of the proposed algorithm
in variable selection and prediction.
We
simulated responses from 11,000 independent data observations total from a normal
population, with a common variance of 2, among which 10,000 observations are
chosen as the training set and the remaining 1,000 observations are used as the
test set. For each of the 11,000 data observations, a vector of 500 predictors
are simulated from normal population with mean 0, and a covariance matrix, . Two
different setup of were used, with the first one being a diagonal
identity matrix, indicating a “perfect” scenario with complete independence
among the predictors; and a second one being a matrix whose entry in row and column is determined by ,
mimicking a “practical” scenario where the nearby predictors are highly
correlated (AR(0.9) correlation structure). Among the 500 predictors, 100 are
randomly chosen to be the “true” signals and three different sets of true
regression coefficients (i.e. the effect size) are chosen as follows to
evaluate the ability of our proposed two-stage algorithm in terms of capturing
the “true” signals when the signals are stronger (i.e., larger effect size) vs.
weaker (i.e., smaller effect size). The remaining 400 predictors are all
associated with zero regression coefficients in the “true” model that generates
the data.
all 100 regression
coefficients are equal to 2;
all 100 regression
coefficients are equal to 0.75;
50% of regression coefficients are
randomly chosen to be 0.75 and the rest are set to be 2.
Let’s summarize what we wish to do with this
simulation study:
How many block should we divide the 10,000 training set observations
into? (4 blocks of 2,500 observation each? 5 blocks? 8,10, 16, 20 or 25 blocks?)
What
is a good threshold for “majority voting?” Analyses of % of the blocks suggest selecting
a variable. %= 50%? 60%? …100%?)
How does the effect size impact the variable selection and prediction?
How does multicolinearity impact the variable selection and prediction?
To
compare the performance of the proposed method under different setups, we
evaluated the following quantities:
Sensitivity (proportion of the “true signals” picked up by the
method, i.e. how many out of the 100 predictors impacting the mean of response
are selected) and specificity (proportions of the 400 “non-signal” predictors
not selected by the method) of variable selection
Mean squared predictor error (MSPE) of the test set.
Here are four graphs
that help us visualize the simulation study findings on the determination of
“majority” when different number of blocks are used.
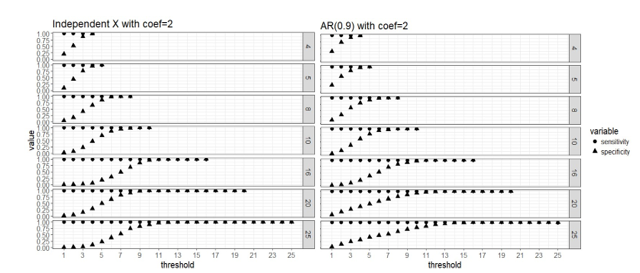
Figure 1. Strong signal
scenario (regression coefficients are all equal to 2): sensitivity and
specificity of the two-stage algorithm when training set are divided into blocks respectively, and a variable is
selected when out of blocks selects this predictor, where =4, 5, 8, 10, 16, 20 and 25, = . Both the case with complete
independence among predictors (left panel) and correlated predictors (right
panel) are presented.
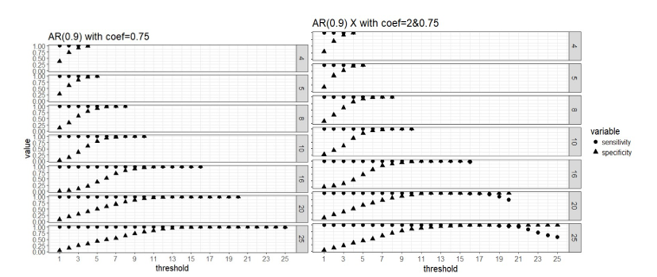
Figure 2. Weak signal
scenario (regression coefficients are all equal to 0.75, left panel) and mixed
signal scenario (regression coefficients are 0.75 or 2, right panel) with
correlated predictors: sensitivity and specificity of the two-stage algorithm
when training set are divided into blocks respectively, and a variable is
selected when out of blocks selects this predictor, where =4, 5, 8, 10, 16, 20 and 25, = .
Apparently that when
parallel computing is possible, it is computationally cheaper to divide the
data into more blocks as there are less observations in each block and the
computation speed is restricted by the computing cost of fitting the model to
each block. Our simulation study did not use a real “BIG” sample size, so the
computing time for each block is not high even when we use as low as 4 block.
But in practice it is possible that we have giant training data and need to
lower the computing cost of fitting a linear regression to a single block
because the blocks still have large samples. All the number of blocks we
considered here seem to approach the same level of sensitivity and specificity
in variable selection, and it seems that 50% or higher blocks agreeing on the
selection of variables is a good threshold for “majority voting.”
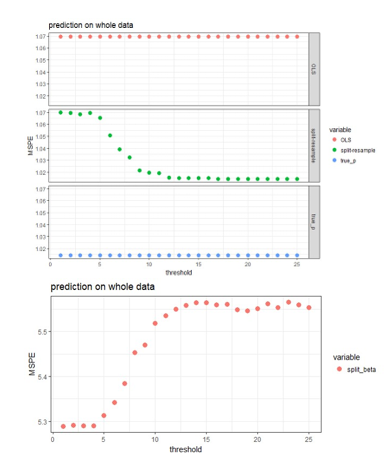
Four approaches are
compared for the case of using 25 blocks on the data with independent predictors
when the effect sizes are 2 (strong signal scenario).
OLS:
fit OLS with all 500 predictors to the original
Split-resample:
fit OLS with selected variables in the proposed approach to the original data
True p: fit OLS with the 100 “true signals” to the original data
Split-resample: predict with aggregated estimation function in Chen and Xie (2014)
Comparison of the
prediction performance can be visualized in the following figures:
Figure 3: MSPE computed
by refitting the model with selected variables to the original data and predict
for the hold out test set.
The aggregated
predictions result in much higher MSPE compared to the other three approaches,
so it is shown in the lower panel of Figure 3, while the other three approaches
are shown in the upper panel of Figure 3. The proposed approaches produces MSPE
that approaches the best scenario when we know exactly which subset of the
predictors are “true signals” in the simulated data when majority voting is
used assuming 50% or higher proportion of blocks is the threshold of selecting
predictors. It seems that the proposed algorithm works well in variable
selection when the predictors are independent or correlated; and also predicts
well when 50% or higher proportion of blocks is the threshold of selecting
predictors. The smaller the effect size, the more blocks need to agree on
variable selection in order to achieve the same sensitivity and specificity,
but this 50% or higher threshold seem to work for both strong and weak signal
case in general.
About the Author
Dr. Jing Zhang is an Associate Professor in the Department of Statistics at Miami University.
After
testing positive for COVID-19 on a rapid antigen test, he missed an opportunity
to meet with the US president who was visiting DeWine’s state. After DeWine was
tested again using a slower, more accurate (RT-PCR) test, he was negative for
COVID-19. A additional test administered a day later also was negative.
Are
there benefits in having a rapid, less accurate test as well as having a
slower, more accurate test? Let’s consider what accuracy means in these tests
and why you might be willing to tolerate different errors at different
times.
I won’t
address how these tests are evaluating different biological endpoints. I’ve
been impressed at how national and local sources have
worked to explain the differences between tests that look for particular
protein segments or for genetic material characteristic of the virus. Richard
Harris (National Public Radio in the US) also provided a nice discussion of
reliability of COVID-19 tests that might be of interest.
I want
to talk about mistakes, errors in testing. No test is perfectly accurate.
Accuracy is good but accuracy can be defined in different ways, particularly in
ways that reflect errors in decisions that are made. Two simple errors are
commonly used when describing screening tests – saying someone has a disease
when, in truth, they don’t (sorry Governor DeWine) or saying someone is disease
free when, in truth, they have the disease. Governor DeWine had 3 COVID-19
tests – the first rapid test was positive, the second and third tests were
negative. Thus, we assume his true health status is disease free.
These
errors are called false positive and false negative errors. (For those of you
who took introductory statistics class in a past life, these errors may have been
labeled differently: false positive error = Type I error and false negative
error = Type II error.) Testing concepts include the complements of
these errors – sensitivity is the probability a test is positive for people
with the disease (1 – false negative error rate) and specificity is the
probability a test is negative for disease-free people (1 – false positive
rate). If error rates are low, sensitivity and specificity are high.
It is
important to recognize these errors can only be made when testing distinct
groups of people. A false positive error only can be made when
testing disease-free people. A false negative error only can
be made when testing people with the disease. An additional challenge is that
the real questions people want to ask are “Do I have the disease if I test
positive?” and “Am I disease free if my test is negative?” Notice these
questions involve the consideration of two other groups –people who test positive
and people who test negative!
Understanding
the probabilities
Probability
calculations can be used to understand the probability of having a disease
given a positive test result — if you know the false positive error rate, the
false negative error rate and the percentage of the population with the
disease, along with testing status of a hypothetical population. The British
Medical Journal (BMJ) provides a nice web calculator for exploring the
probability that a randomly selected person from a population has the disease
for different test characteristics. In addition, the app interprets the
probabilities in terms of counts of individuals from a hypothetical population
of 100 people classified into 4 groups based upon true disease status (disease,
no disease) and screening test result (positive, negative).
It is
worth noting that these probabilities are rarely (if ever) known and can be
very hard to estimate – particularly when changing. In real life,
there are serious challenges in estimating the numbers that we get fed into
calculators such as this – but that’s beyond scope of this
post. Regardless, it is fun and educational to play around with the
calculator to understand how things work.
These
error rates vary between different test types and even for tests of the same
type. One challenge that I had in writing this blog post was obtaining error
rates for these different tests. Richard Harris (NPR) reported
that PCR false positives from the PCR test were approximately 2%, with
variation attributable to the laboratory conducting the study and the
test. National Public Radio reported
that one rapid COVID-19 test had a false negative error rate of approximately
15% while better tests have false negative tests less than 3%. One
complicating factor is that error rates appear to depend on when the test
is given in the course of disease.
Examples
The
following examples illustrate a comparison of tests with different accuracies
in communities with different disease prevalence.
Community
with low rate of infection
A
recent story about testing in my local paper reported 1.4% to 1.8% of donors to
the American Red Cross had COVID-19. Considering a hypothetical population with
100 people, only 2 people in the population would have the disease and 98 would
be disease free.
Rapid,
less accurate test: Suppose we have a rapid test with a 10% false positive
error rate (90% specificity), 15% false negative error rate (85% sensitivity)
and 2% of people tested are truly positive. With these error rates, suppose
both of the people with the disease test positive and 10 of the 98 disease-free
people test positive. Based on this, a person with a positive test (2 + 10= 12)
has about a 16% (2/12 x 100) chance of having the disease, absent any other information
about exposure.
Disease
No Disease
Total
Test +
2
10
(98 x .10)
12
2/12
(16%)
Test –
0
(2 x .15)
88
89
Total
2
98
100
For a hypothetical population of 100
people with 2% infected, a false positive rate of 0.10, and a false negative
rate of 0.15, the chance of having the disease given a positive test is about
16%.
Slower,
more accurate test: Now, suppose we have a more accurate test with a 2% false
positive error rate (98% specificity) and 1% false negative error rate (99%
sensitivity). With these error rates, both of the people with the disease test
positive and 2 of the 98 disease-free people test positive. Based on this, a
person with a positive test (2 + 2= 4) has about a 50% (2/4) chance of having
the disease.
Disease
No Disease
Total
Test +
2
2
(98 x .02)
4
2/4
(50%)
Test –
0
(2 x .01)
96
96
Total
2
98
100
For a hypothetical population of 100
people with 2% infected, a false positive rate of 0.02, and a false negative
rate of 0.01, the chance of having the disease given a positive test is about
50%.
Community
with a higher rate of infection
Now
suppose we test in a community where 20% have the disease. Here, 20 people in
the hypothetical population of 100 have the disease and 80 are disease free.
This 20% was based on a different news source suggesting that 20% was one of
the highest proportions of COVID-19 in a community in the US.
Rapid,
less accurate test: Consider what happens we use a rapid test with a 10%
false positive error rate (90% specificity) and 15% false negative error rate
(85% sensitivity) in this population. With the error rates described for this
test, 17 of the 20 people with disease test positive and 8 of the 80
disease-free people test positive. Based on this, a person with a positive test
(17 + 8 = 25) has about a 68% (17/25) chance of having the disease without any
additional information about exposure.
Disease
No Disease
Total
Test +
17
8
(80 x .10)
25
17/25
(68%)
Test –
3
(20 x .15)
72
75
Total
20
80
100
For a hypothetical population of 100
people with 20% infected, a false positive rate of 0.10, and a false negative
rate of 0.15, the chance of having the disease given a positive test is about
68%.
Slower,
more accurate test: Now suppose we apply a more accurate test with a 2% false
positive error rate (98% specificity) and 1% false negative error rate (99%
sensitivity) to the same population. In this case, all 20 people with the
disease test positive and 2 of the 80 disease-free people test positive. Based
on this, a person with a positive test (20 + 2 = 22) has about a 90% (20/22)
chance of having the disease.
Disease
No Disease
Total
Test +
20
2
(80 x .02)
22
20/22
(90%)
Test –
0
(20 x .01)
78
78
Total
20
80
100
For a hypothetical population of 100
people with 20% infected, a false positive rate of 0.02, and a false negative
rate of 0.01, the chance of having the disease given a positive test is about
90%.
Returning
to the big question
So, if
you test positive for COVID-19, do you have it? If you live in a community with
little disease and use a less accurate rapid test, then you may only have a 1
in 6 chance (16%) of having the disease (absent any additional information
about exposure). If you have a more accurate test, then the same test result
may be associated with a 50-50 chance of having the disease. Here, you might
want to have a more accurate follow up test if you test positive on the rapid,
less accurate test. If you live in a community with more people who
have the disease, both tests suggest you are more likely than not to have the
disease. Recognize that these tests are being applied in situations with
additional information being available including whether people exhibit
COVID-19 symptoms and/or live or work in communities with others who have
tested positive.
Final
thoughts
You
might be interested in controlling different kinds of errors with different
tests. If you are screening for COVID-19, you might want to minimize false
negative errors and accept potentially higher false positive error rates. A
false positive error means a healthy disease-free person is quarantined and
unnecessarily removed from exposing others. A false negative error means a
person with disease is free to mix in the population and infect others. So,
does Governor DeWine have COVID-19? Ultimately, the probability that the
governor is disease-free reflects the chance of being disease-free given one
positive result on a less accurate test and two negative results from more
accurate tests. The probability he is disease-free is very close to one, given
no other information about exposure.
About the Author
Dr. A. John Bailer is a University Distinguished Professor and Chair in the Department of Statistics at Miami University.